






In This Article
When we get a prescription from a doctor, the drug usually comes with instructions for how to use it. These include when we take the drug (before or after a meal), how many times a day and for how long we take it, and what we have to avoid while using the drug. Doctors also ask if we use any other medication or if we have any chronic disease before prescribing any new medication. Most medicines look similar, but drugs themselves can be very different. Have you thought about what happens to a drug when it gets into our body?
All body functions and disease processes—as well as most drug reactions—occur at the cellular level. Drugs are chemicals that alter basic processes in body cells. They cannot add functions and activities but can only affect normal cellular functions and activities. Drugs given for systemic effects must reach adequate concentrations in the blood and other tissue fluids surrounding our cells to have an effect. Drugs must circulate through their body, reach their intended cells, and, after affecting the cells, be eliminated from the body. But how do all these processes occur? How do systemic drugs reach, interact with, and leave the body’s cells?
The answers to these questions are answered by one of the main domains of pharmacology—pharmacokinetics. Pharmacokinetics is derived from the Greek words pharmakon (drug) and kinetikos (movement) and means “what the body does to a drug.” It refers to the movement of a drug into, through, and out of the body.
Knowledge of pharmacokinetic principles helps healthcare providers to adjust dosages more accurately and rapidly. Application of pharmacokinetic principles to individualize pharmacotherapy is termed therapeutic drug monitoring and aims to give the exact amount of drug to each person.
The four main parts of the pharmacokinetic processes are
- Absorption
- Distribution
- Metabolism
- Elimination
These processes are abbreviated as ADME.
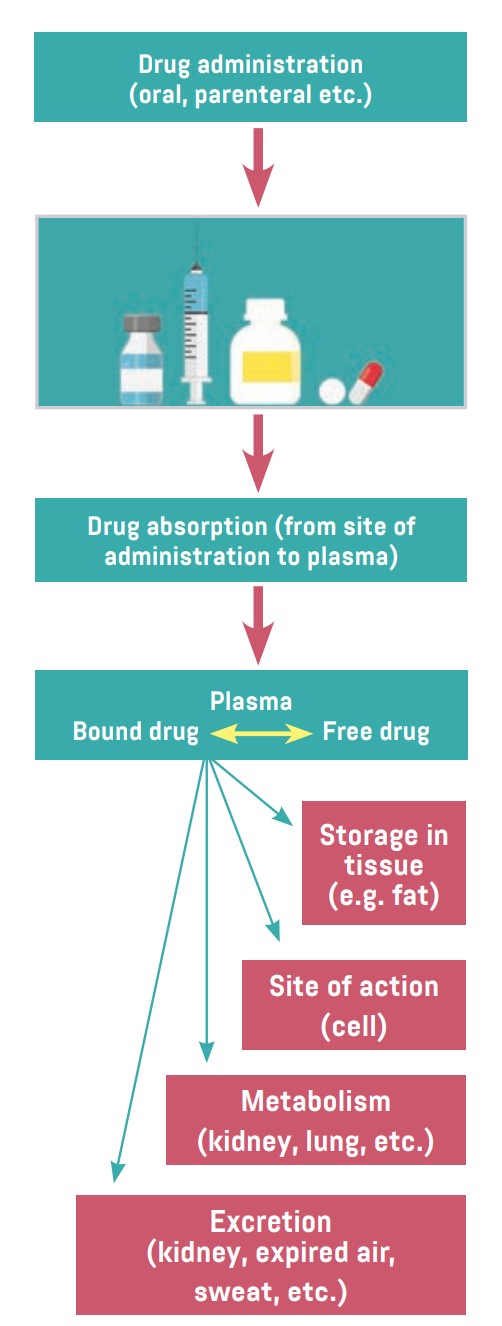
Figure. Entry and movement of drug molecules through the body to sites of action, as well as metabolism and excretion
Absorption
Absorption is the process that occurs when a drug enters the body until it enters the bloodstream to be circulated. The duration of a drug’s action is largely determined by the rate of absorption, and intensity is determined by the extent of absorption.
Numerous factors affect the rate and extent of drug absorption, including dosage, form, route of administration, blood flow to the site of administration, gastrointestinal function, and the presence of food or other drugs.
Most oral drugs must be swallowed, dissolved in gastric fluid, and delivered to the small intestine (which has a large surface area for absorption of nutrients and drugs) before they are absorbed. Liquid medications are absorbed faster than tablets or capsules because they need not be dissolved. Rapid movement through the stomach and small intestine may increase drug absorption by promoting contact with absorptive mucous membrane; it also may decrease absorption because some drugs may move through the small intestine too rapidly to be absorbed. For many drugs, the presence of food in the stomach slows the rate of absorption and may decrease the amount of drug absorbed. That is why some drugs are taken before meals and some after.
Drugs cannot be absorbed well when a patient has diarrhea and the intestines are working excessively; the effect of the drug will be reduced. Absorption may be increased if the patient has constipation and the intestines are working very slowly; this could have unforeseen adverse effects.
Drugs injected into subcutaneous (under the skin) or intramuscular (in the muscles) tissues are usually absorbed more rapidly than oral drugs because they move directly from the injection site to the bloodstream. Absorption is rapid from muscle sites because muscle tissue has abundant blood supply. Drugs injected intravenously (into the veins) do not need to be absorbed because they are placed directly into the bloodstream.
Other absorptive sites include the skin, mucous membranes, and lungs. Most drugs applied to the skin are given for local effects (e.g., muscle relaxant creams, itch relief ointments, and burn ointments). Systemic absorption is minimal from intact skin but may be considerable when the skin is inflamed or damaged. Also, a number of drugs have been formulated in adhesive skin patches for absorption through the skin. Some drugs applied to mucous membranes also are given for local effects. However, systemic absorption occurs from the mucosa of the oral cavity, nose, eyes, etc. Drugs absorbed through mucous membranes pass directly into the bloodstream. For example, nitroglycerin sublingual tablets are used to treat episodes of angina (chest pain) in people who have coronary artery disease. It is also used just before activities that may cause episodes of angina to prevent symptoms. It works by relaxing the blood vessels so the heart does not need to work as hard and therefore does not need as much oxygen. The lungs can be a good absorption site, as they have a large surface area for absorption of anesthetic gases and a few other drugs.
Distribution
Distribution involves the transport of drug molecules within the body. Once a drug is injected or absorbed into the bloodstream, it is carried by the blood and tissue fluids to its sites of pharmacologic action, metabolism, and excretion. Most drug molecules enter and leave the bloodstream at the capillary level, through gaps between the cells that form capillary walls.
Distribution depends largely on the adequacy of blood circulation. Drugs are distributed rapidly to organs receiving a large blood supply, such as the heart, liver, and kidneys. Distribution to other internal organs, muscle, fat, and skin is usually slower.
An important factor in drug distribution is protein binding. Most drugs form a complex with plasma proteins such as albumin. These proteins act as carriers for the drugs. Drug molecules bound to plasma proteins are pharmacologically inactive because the large size of the complex prevents their leaving the bloodstream through the small openings in capillary walls and reaching their sites of action. Only the free or unbound portion of a drug acts on the body’s cells. As the free drug acts on cells, the decrease in plasma protein levels causes some of the bound drug to be released.
Protein binding allows part of a drug dose to be stored and released as needed. Some drugs also are stored in muscle, fat, or other body tissues and released gradually when plasma drug levels fall. These storage mechanisms reduce the risk of toxicity. Drugs that are highly bound to plasma proteins or stored extensively in other tissues have a long duration of action. This affects the frequency of intake of a drug and is why some drugs are taken more frequently.
Drug distribution into the central nervous system (CNS) is limited because of the blood–brain barrier, which is composed of capillaries with tight walls and limits the movement of drug molecules into brain tissue. This barrier has been created to act as a selectively permeable membrane to protect the CNS. However, it also can make drug therapy for CNS disorders more difficult because drugs must pass through the cells of the capillary wall rather than between cells. As a result, only drugs that are lipid soluble or have a transport system can cross the blood–brain barrier and reach therapeutic concentrations in brain tissue.
Drug distribution during pregnancy and lactation is also unique. During pregnancy, most drugs cross the placenta and may affect the fetus. During lactation, many drugs enter breast milk and may affect the nursing infant. This barrier is also important and protects the fetus from the adverse effects of drugs.
If we have valuable things at home or at work, we put them in extra safe places and use extra security systems to protect them. So it is with our body: our brain and a fetus are extra protected by these barriers.
Metabolism
Metabolism is the chemical reaction by which drugs are inactivated or bio-transformed by the body. Most often, an active drug is changed into one or more inactive metabolites, which are then excreted. Some active drugs yield metabolites that are also active and that continue to exert their effects on the body’s cells until they are metabolized further or excreted. Other drugs (called prodrugs) are initially inactive and exert no pharmacologic effects until they are metabolized.
Most drugs are lipid soluble, a characteristic that aids their movement across cell membranes. However, the kidneys, which are the primary excretory organs, can excrete only water-soluble substances. Therefore, one function of metabolism is to convert fat-soluble drugs into water-soluble metabolites.
Hepatic drug metabolism—or clearance—is a major mechanism for terminating drug action and eliminating drug molecules from the body. Most drugs are metabolized by enzymes in the liver, red blood cells, plasma, kidneys, lungs, and gastrointestinal mucosa.
With long term administration, some drugs stimulate liver cells to produce larger amounts of drug metabolizing enzymes (a process called enzyme induction). Enzyme induction accelerates drug metabolism because larger amounts of the enzymes allow larger amounts of a drug to be metabolized during a given time. As a result, larger doses of the rapidly metabolized drug may be required to produce or maintain therapeutic effects. Metabolism also can be decreased or delayed in a process called enzyme inhibition, which most often occurs with concurrent administration of two or more drugs that compete for the same metabolizing enzymes. In this case, smaller doses of the slowly metabolized drug may be needed to avoid adverse reactions and toxicity from drug accumulation. Thus, doctors always ask about any medications already being used before prescribing a new medication. Also, some foods (eg. grapefruit), drinks (alcohol), and smoking affect the activity of the enzymes which are metabolizing the drugs in the liver. Thus, we must be careful and follow a doctor’s instructions while using any medication.
The rate of drug metabolism is also reduced in infants (their hepatic enzyme system is immature), in people with impaired blood flow to the liver or severe hepatic or cardiovascular disease, and in people who are malnourished or on low-protein diets. Metabolism in elderly people and pregnant women also changes a lot; thus, the dose given to each individual should be carefully calculated—especially in children, the elderly, and pregnant women.
Excretion
Excretion refers to the elimination of a drug from the body. Effective excretion requires adequate functioning of the circulatory system and of the organs of excretion (kidneys, bowel, lungs, and skin). Most drugs are excreted by the kidneys and eliminated unchanged or as metabolites in urine. Some drugs or metabolites are excreted in bile, then eliminated in feces; others are excreted in bile, reabsorbed from the small intestine, returned to the liver (called enterohepatic recirculation), metabolized, and eventually excreted in urine. Some oral drugs are not absorbed and are excreted in the feces. The lungs mainly remove volatile substances, such as anesthetic gases. The skin has minimal excretory function. Factors impairing excretion, especially severe renal disease, lead to accumulation of numerous drugs and may cause severe adverse effects if dosage is not reduced. Doctors always ask about any chronic diseases before prescribing any medication.
The process of developing a drug is long and difficult. The instructions for usage are decided after many experimental and clinical trials. None of the instructions given with medications are decided randomly; thus, we must completely follow the instructions to maximize the positive effects of a drug and to avoid any possible adverse effects.
References
- Abrams, Anne Collins., Carol Barnett Lammon, and Sandra Smith Pennington. Clinical Drug Therapy: Rationales for Nursing Practice. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2007.
- Basic&Clinical Pharmacology, 12th Edition, McGrawHill Lange.
- Modern Pharmacology with Clinical Applications, Sixth Edition, Charles R. Craig and Robert E. Stitzel, Lippincott Williams &Wilkins.



